Assessment of the potential vector threat of bat-borne pathogens and the host-associated ectoparasites in the provinces of Vientiane and Khammouane of the Lao PDR (BatMap Project Extension)
Funded by the U.S. Naval Medical Research Center-Asia (NMRC-A) in support of the Department of Defense Global Emerging Infections Surveillance and Response System (DoD-GEIS)

Project Coordinator:
1. Dr. Paul Brey, Director, Institut Pasteur du Laos, Vientiane, Lao PDR
2. Dr. Ian Sutherland, Chief of Entomological Sciences, U.S. Naval Medical Research Center – Asia, Sembawang, Singapore
3. Dr. Jeffrey Hertz, Chief of Entomological Sciences, U.S. Naval Medical Research Center – Asia, Sembawang, Singapore
Members of staff in Institut Pasteur du Laos:
1. Khamsing Vongphayloth
2. Khaithong Lakeomany
3. Nothasine Phommavanh
Background
The biological and social attributes of bats make them ideal reservoirs for emerging infectious diseases (EIDs). Bats represent the largest number within mammalian species (>925), occupy a variety of ecological niches, roost in large numbers, and are able to disperse over wide geographical distances. Until now, no reference data has been available regarding the viral and pathogen make-up of bats, their ectoparasitic fauna, and their potential to be an EID source within Laos. The growing US force presence in this region and the central geostrategic positioning of Laos in the Indochinese peninsula (sharing borders with China, Myanmar, Vietnam, Cambo dia, and Thailand) justify the urgent need to establish an inventory of the geographic distribution of bat species, of their ectoparasites, and of the pathogens bred and potentially transmitted by both bats and/or their ectoparasites.
Vector-borne diseases, including bat-borne agents, constitute a significant infectious disease risk for local populations. In Laos, definitive diagnosis is often not available for vector-borne illnesses, thus infectious diseases that are a threat to military and civilian populations remain misdiagnosed and unreported. In order to identify common and emerging vector-borne pathogens in Laos, NMRC-A Singapore (SG) has established a study to assess the distribution and infection potential of bat ectoparasites and bat-borne pathogens. In this study, bat ectoparasites and associated arthropod vectors were surveyed to provide biological specimens for diagnostic purposes. The samples were transported to the Institut Pasteur du Laos (IPL) laboratory in Vientiane, where a wide range of diagnostic tests were performed to identify both the vector and the pathogens with which they may be infected. This study is known as the “Vector mapping of bat-borne ectoparasites and pathogens in the Lao PDR” (BatMap).
In 2017 annual report, we updated our activities of the project including field collection, identification, extraction, and development of molecular products for Lao repository, and reference for future studies. In this report in 2018, we updated our project activities extending from 2017, focusing on pathogen detection by our partners.
Objectives
+ Obj 1: help address the knowledge gap in bat-borne virus and ectoparasite biology, identification, and distribution in Vientiane and Khammouane provinces.
+ Obj 2: conduct molecular biology detection studies for a panel of viruses and bacteria in collected specimens.
+ Obj 3: provide/improve infectious disease surveillance competencies (building of local capacity and competencies).
Methods
Field site collection
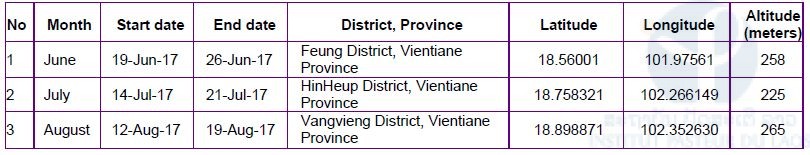
Due to administrative/authorization delays, our first field collection of bats was carried out late, in June (Q4 of the project). In collaboration with the Faculty of Environmental Sciences, National University of Laos, three field missions have been completed in three districts of Vientiane Province (Figure 1, Table 1). However, because of this delay, it was not possible to carry out any field missions in Khammouane Province.
Figure 1. Map of field sites (red area) in Vientiane Province
Table 1: Field site collection
Animal capture for research statement
The bat study was approved by the Institutional Animal Care and Use Committee (IACUC) (Protocol number: EXM-17-15-NAMRU-2). For the Lao side, the authorization agreement for carrying out bat capture was approved by the wildlife authorities of the Department of Forest Resource Management (DFRM), and the Ministry of Agriculture and Forestry, Lao PDR, No.0174/DFRM, issued on January 2017.
Bat collection and identification procedure

Figure 2. Harp traps and mist nets setting
Bats were collected using four-bank harp traps (Francis, 1989) and mist nets (Figure 2). Harp traps were set across natural trails and over small streams in the forest understorey, and at the entrance of caves in relatively concealed conditions Mist nets were also set at similar places to harp traps, but were more often used in open spaces. Both harp traps and mist nets were set before sunset; harp nets were left overnight, mist nets were taken down around 2200.
Captured bats were held individually in cloth bags. Species, sex, age (adult or juvenile), and reproductive condition (pregnant or lactating) were determined in the field. Species were identified following Francis (2008), Csorba et al. (2003), and Corbet and Hill (1992). Adults or juveniles were identified by the presence of unfused epiphyses of the phalanges and metacarpal joints (Brunet-Rossinni and Wilkinson, 2009). The reproductive status of female bats was determined by examining the nipples (Racey, 2009). Some external characteristics, including forearm length (FA), were measured using calipers and body mass (W) was taken using a Pesola spring balance. Most bats were marked with wing bands for individual identification and were released at the capture point within 12 hours.
For each species, two or three specimens were collected to confirm identification. Bats were euthanized using chloroform. Specimens were fixed in 95% ethanol in the field and were transferred to 70% ethanol when they were brought back to the laboratory. All specimens were registered and catalogued in the Zoological Collection of the Faculty of Environmental Sciences, National University of Laos, Lao PDR.
Bat sampling procedure
Live bats were placed into porous cotton bags (with draw-string mouths) and kept in a cool dry place for six hours or less until sampling time. The following basic set of samples were collected from each bat: (i) Saliva (oropharyngeal swab): 1 aliquot; (ii) Feces (fresh fecal sample or rectal swab): 1 aliquot; (iii) Blood: 1 aliquot; (iv) Urine (urogenital swab): 1 aliquot; and (v) Ectoparasites : 1 aliquot (Figure 3). The whole blood (serum; rbc/wbc pellet) was diluted by 1/10 of PBS 1x, centrifuged by mini-centrifuge machine, and then divided into two aliquots (serum and blood cells). Ectoparasites were collected using flame-sterilized forceps. All samples were stored at −20°C, then transported to IP-Laos in Vientiane and stored at −80°C until laboratory analysis.
Figure 3. Bat sample sampling

Lab activity
This study focused on some genera of arbovirus and non-arbovirus which previous studies had commonly found in bats, such as flavivirus, alphavirus, phlebovirus, and filovirus, by using appropriate techniques to detect each viral genus.
Extraction
Total nucleic acids (DNA/RNA) were extracted from bio-samples (blood samples and ectoparasites) of bats by using Nuclo Spin®8 Virus (MACHEREY-NAGAL, Germany) in an extraction room.
Lab techniques and assay procedure for Arbovirus genus screeni ng at IP-Laos
Multiple sets of primers have been selected and tested for screening specimens for the presence of phlebovirus, flavivirus, and alphavirus sequences by means of conventional nested RT-nPCR as previously described by Sanchez-Seco et al.(Sanchez-Seco, Rosario et al. 2001; Sanchez-Seco, Rosario et al. 2005). Amplicons were analyzed by agarose gel electrophoresis. Positive controls were included in all series using RNA extracted from culture supernatants of reference strains (i) for Alphavirus: Sindbis, Chikungunya, and Me Tri, (ii) Phlebovirus: strain IPL-13-214, and (iii) Flavivirus: dengue and Japanese encephalitis.
Next generation sequencing for identification of viruses in bat mites and batflies
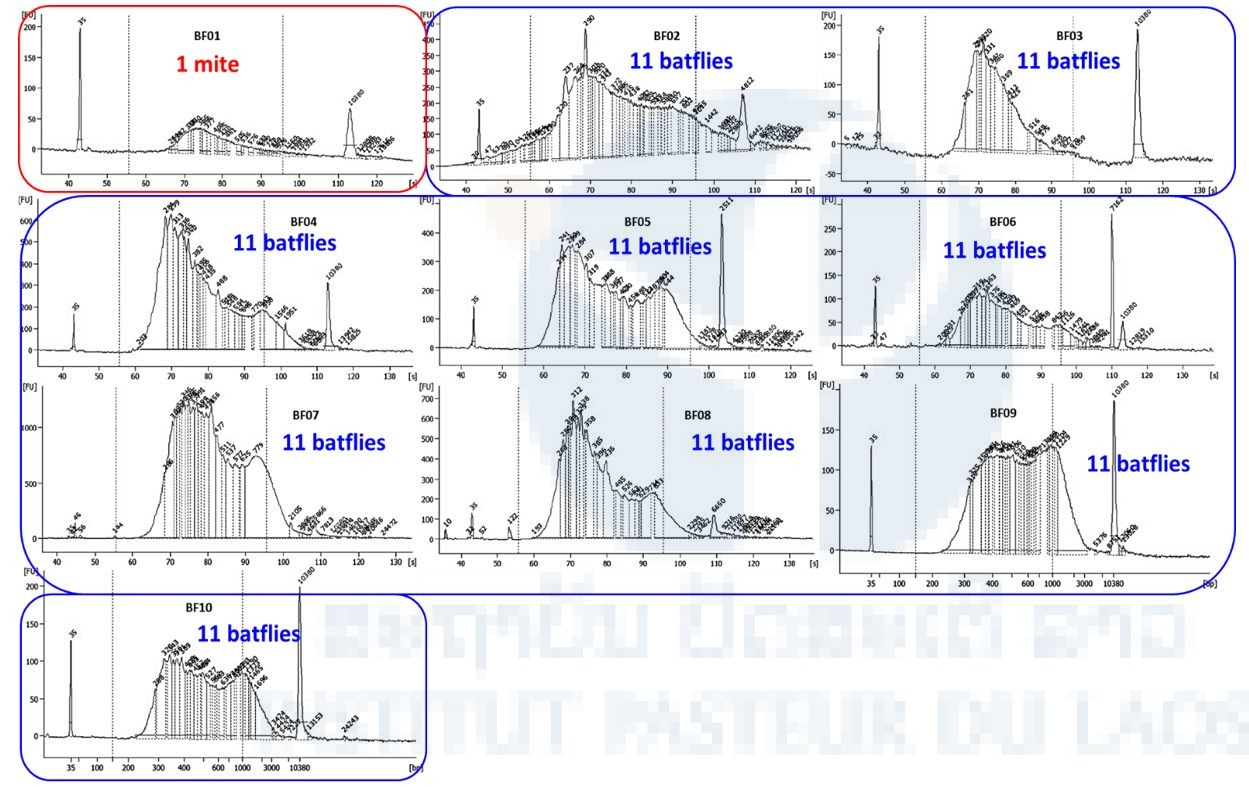
The NGS sequencing of one mite and 99 batflies collected on Laotian bats was performed by the Pathogen Discovery team of the Institut Pasteur in Paris. Starting from the total nucleic acids extracted in IP Laos, ten libraries were constructed using the SMARTer® Stranded Total RNA-Seq Kit v2-Pico Input Mammalian (Clontech). Size and quality of the libraries were checked on an Agilent bioanalyzer (Figure 4) before being sequenced on a NextSeq 500 sequencer (Illumina) and a NextSeq® 500/550 High Output Kit v2 (300 cycles) flow cell.
Figure 4. BioAnalyzer profile of the 10 libraries.
Bacterial screening
The bacterial screening was carried out in collaboration with the Lao-Oxford University-Mahosot Hospital-Wellcome Trust Research Unit (LOMWRU), based at Mahosot Hospital, Vientiane. To investigate the occurrence of Rickettsia spp., Leptospira spp., and Bartonella spp. in bats and bat ectoparasites in Laos, a molecular screening approach was taken using LOMWRU’s protocols.
Results
Bat capture
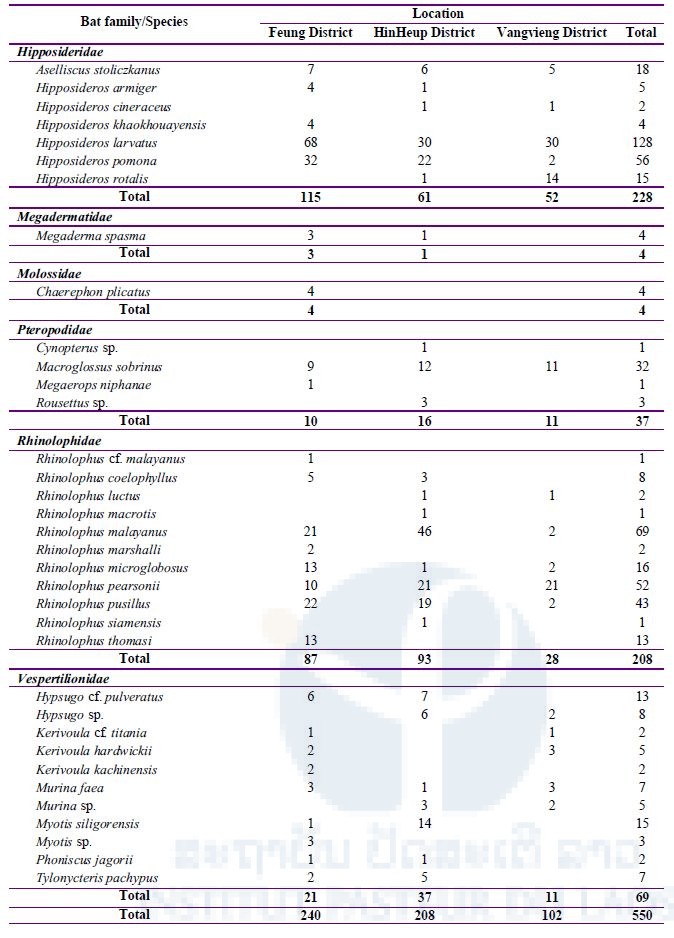
A total of 550 bats were captured from three districts in Vientiane Province during our three missions. Bats were classified into 35 species (Table 2); a total of 26 species (240 bats) were collected from Feung District, 25 species (208 bats) from HinHeup District, and 16 species (102 bats) from Vangvieng District.
Table 2: Total number of bats captured during three field missions in three districts, Vientiane Province
Bat sample sampling
Bat biological samples
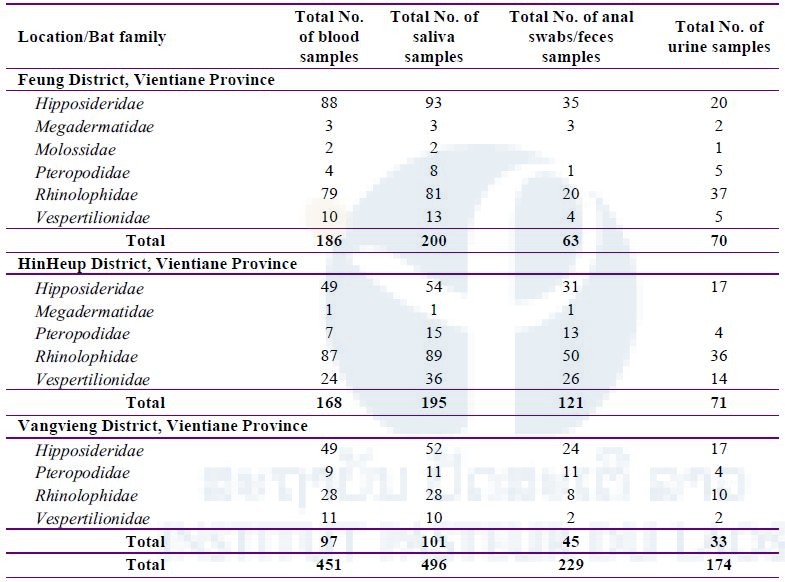
A total of 496 saliva, 451 blood, 229 anal swabs/feces, and 174 urine samples were taken from 498 bats belonging to 6 families: Hipposideridae, Megadermatidae, Molossidae, Pteropodidae, Rhinolophidae, and Vespertilionidae (Table 3).
Table 3: Total number of bat samples (saliva, blood, anal swabs/feces, and urine) collected during three field missions in three districts, Vientiane Province
Bat ectoparasites
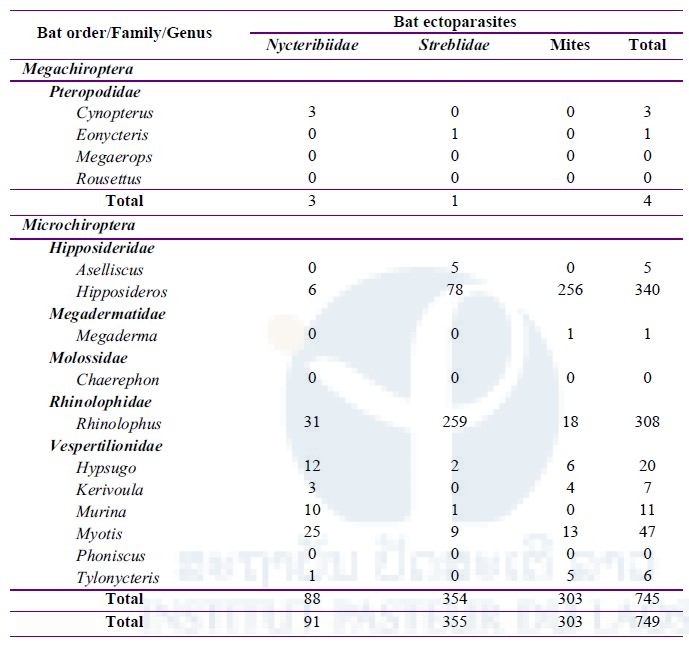
From 550 bats captured and examined for ectoparasites (Table 2 above), a total of 749 ectoparasites were collected, divided between bat mites (303) and two families of batflies (Figure 5): Nycteribiidae (91) and Streblidae (355) (Table 4 below).

Figure 5. Batflies and bat mites
Table 4: Total number of ectoparasites (batflies and mites) collected during three field missions in three districts, Vientiane Province
Arboviral screening
Arboviral detection in bat blood samples
A total of 287 bat blood samples, belonging to 6 bat families—Hipposideridae, Megadermatidae, Molossidae, Pteropodidae, Rhinolophidae, and Vespertilionnidae—were screened for arboviruses using RT-nested PCR techniques for the presence of arbovirus sequences (pan flavi, pan alpha, and pan phlebo). Of 287 blood samples, alphaviruses were detected in 25 samples (8.71%), all collected from Hinheup District. Flavivirus sequences were detected in 13 samples (4.53%), all also collected from Hinheup District. Phlebovirus RNA was detected in 11 bats (3.83%), of which 9 were Hipposideridae (8 collected from HinHeup District and 1 from Fueng District) and 2 were Rhinolophidae collected from Feung District.
Arboviral detection in bat ectoparasites
A total of 288 pools of 468 batflies were screened for pan flavi, pan alpha, and pan phlebo. Overall, 10/288 pools (3.47%) were positive for alphaviruses—8/10 (80%) were Streblidae, 1/10 (10%) were Nycteribiidae, and 1/10 (10%) were Spinturnicidae. Flaviviruses were detected in 3/288 pools (1.04%), of which 2/3 (66.67%) was detected from Spinturnicidae, and 1/3 (33.33%) from Nycteribiidae. Only 1/288 pools (0.35%) of Streblidae was positive for phleboviruses.
Next-generation sequencing for identification of viruses in bat mites and batflies
We obtained 396,445,467 paired reads of 150 pb each in total for the ten libraries. The repartition of raw reads for each library is presented in Table 5 (i.e. nb clusters). Except for pool n°1 (mite) for which we obtained only 5.7 million paired reads, we obtained 24 to 55 million paired reads for each of the 9 pools of batflies. After quality check and trimming using the Galaxy portal, cleaned reads were assembled into 121,592 to 2,172,370 contigs in function of the sample. The remaining non-assembled reads (singletons, up to 2 million sequences) and contigs were then translated using an in-house ORF prediction tool. Taxonomic assignation of the sequences was obtained by BlastP against the curated comprehensive rVDB viral database developed and maintained by our team (https://hive.biochemistry.gwu.edu/rvdb) with an E-value cut-off of 10-3; viral hits were further confirmed or infirmed by comparison of the blast score obtained after BlastP against the whole NCBI nr database.
The bioIT analysis of bacteria and eukaryote microorganisms present in the different arthropods is still ongoing.
Validated viral sequences are presented in Table 6 according to their taxonomic assignation. Several viruses belonging to the Rhabdoviridae, Chuviridae (a proposed viral family within the Mononegavirales order), the Bunyavirales and Picornavirales orders, Reoviridae, Orthomyxoviridae, Retroviridae, Flaviviridae, and Parvoviridae were detected in the different samples. In Table 6a, viruses whose genomes are close to previously published sequences (>90% identity at the nucleic acid level in the polymerase gene) are highlighted in yellow, while viruses with a sequence more distant at the nucleic acid level are presented in grey. Viral genomes were differently covered by the sequencing, depending on the initial load of the samples. In Table 6b, green cells represent a coverage >50% of the closest viral genome; orange cells represent a medium coverage (10–50%) and red cells represent a limited coverage(<10%).
Follow-up studies at the Institut Pasteur Paris (Dr. Marc Eloit, Pathogen Discovery Laboratory)
After validation of viral sequences, follow-up studies are needed to further confirm the results of bioIT analyses and the identification of viral sequences: 1) Mapping of the sequences on close reference genomes; 2) Confirmation of the presence of viral genome in the RNA extracts by PCR or qPCR; 3) Depooling to identify the individual ectoparasites involved.
Flaviviridae-related sequences
An unexpected finding was the identification of West Nile virus sequences, a Flavivirus known to be transmitted by mosquitoes, in one mite pool sample (BF01) and one batfly pool sample (BF07). Mapping of these sequences is shown on Figure 6a, and the coverage and nucleotide identity with the WN virus of this virus in Table 6. Preliminary phylogenetic analyses surprisingly clustered this WN with a strain isolated in Israel. Several attempts to amplify various portions of the WN genome were performed, both on the pool cDNA and on the individual extracts, and also on the NGS libraries, with different PCR and qPCR systems, without any positive results. After verification with other users of the sequencer, it has been noted that the previous run of sequencing was a supernatant viral culture of the same WN strain, and that this previous run used the same barcodes as the one we used, possibly explaining why these previous WN sequences were assigned to our run. All these observations result in interpreting these WN sequences as a cross-contamination from the previous NGS run performed in the sequencer.
Bunyavirales-related sequences
Orthobunyavirus-related sequences were identified in BF08 and were close to Wolkberg virus, a novel Orthobunyavirus recently reported in batflies from South Africa (Jansen van Vuren et al.) related to two bat viruses (Mojuí dos Campos and Kaeng Khoi viruses) and to an arbovirus (Nyando virus) previously shown to infect humans. This virus presents a genome coverage of approximately 78% (Figure 6b) and a nucleic identity of 74% in the polymerase gene with the reported Wolkberg virus. The presence of viral genome in the initial pool was tested by qPCR on the cDNA of the pool and individual samples. One batfly (E167) gave an amplicon at the expected size. Confirmation by Sanger sequencing is ongoing.
Mononegavirales-related sequences
Rhabdoviridae- and Chuviridae- (a proposed viral family within the Mononegavirales order) related sequences were identified in four pools of batflies (Table 6), with different coverage levels (Table 6b) and rather distant from the closest reference sequences (Table 6a). Confirmation of the presence of viral genomes in the initial RNA pools and identification of positive individual extracts is ongoing.
Orthomyxoviridae-related sequences
Quaranjavirus-related sequences were identified in five pools of batflies, with different coverage levels (Table 6b). For example, virus identified in the BF08 sample covered less than 15% of the genome of the closest virus, but with sequences obtained in at least three segments of the virus (Figure 6c), while the BF04 sample is nearly covered at 24% of the genome. Confirmation of the presence of viral genomes in the initial RNA pools and identification of positive individual extracts is ongoing.
Other viral sequences
Sequences related to the Picornavirales order and to the Reoviridae, Retroviridae, and Parvoviridae were also identified in the different pools, with various levels of genome coverage but all rather distant from the closest reference genomes. Confirmation of the presence of viral genomes in the initial RNA pools and identification of positive individual extracts will be considered in function of available resources.
Table 5: Metrics of the run
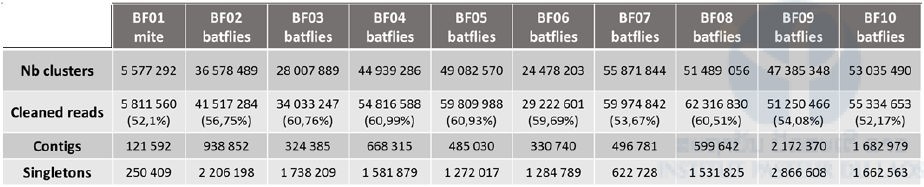
Table 6: Main viruses identified in the 10 libraries and their corresponding coverage scale and genetic distances to the closest viral genome.
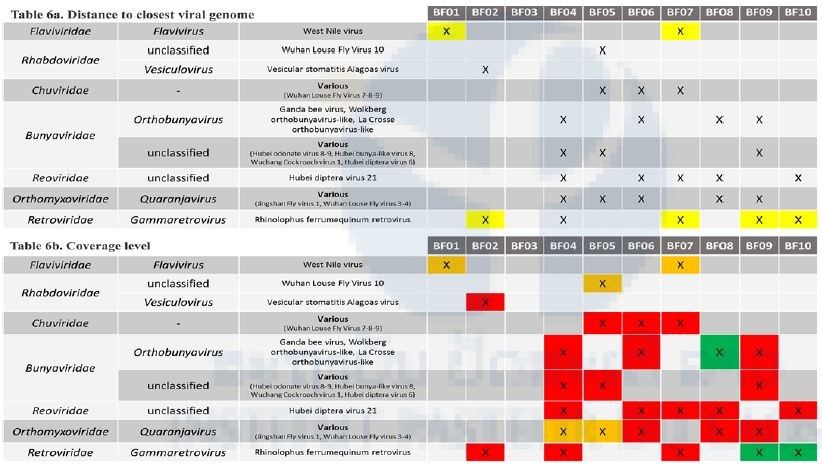
In 6b., green cells represent a coverage >50% of the closest viral genome; orange cells represent a medium coverage (10-50%) and red cells represent a limited coverage (<10%)
Figure 6. Examples of mapping of viral sequences identified in batflies datasets.
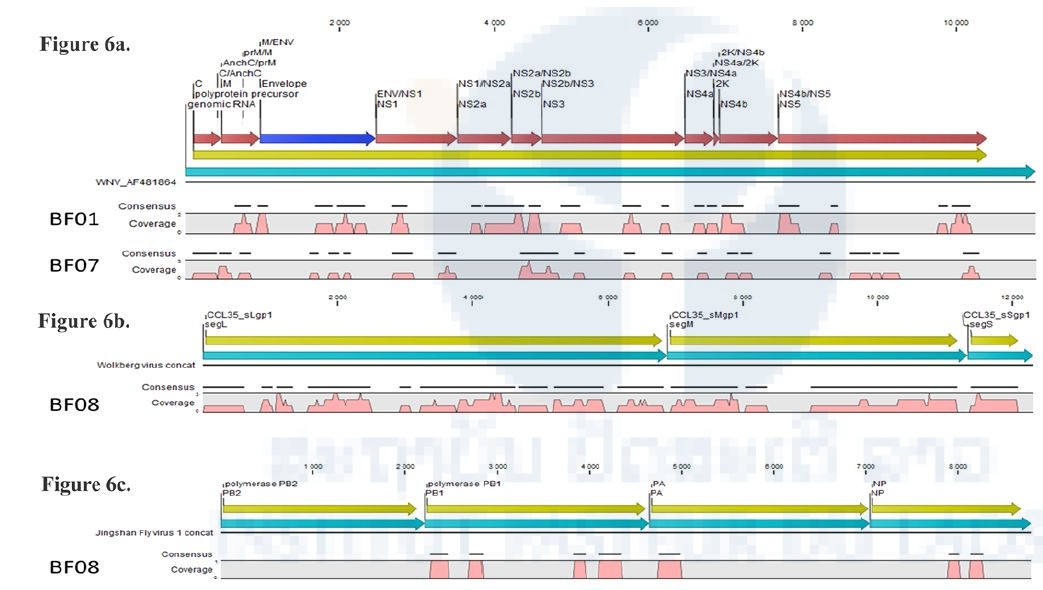
Pink represents the coverage of the genome, yellow the coding sequences of the reference viral genome. When the closest viral genome was segmented, sequences were concatenated for better clarity.
Bacterial screening
Bat blood samples-bacterial screening
A total of 265 bat blood samples, belonging to 5 bat families—Hipposideridae, Megadermatidae, Pteropodidae, Rhinolophidae, and Vespertilionnidae—were screened for Rickettsia spp., Leptospira spp., and Bartonella spp. Of 265 blood samples, 26 samples (9.81%) were positive for Rickettsia spp.: 14/26 (53.85%) Rhinolophidae, 7/26 (26.92% ) Hipposideridae, 3/26 (11.54%), Vespertilionnidae, and 2/26 (7.69%) Pteropodidae. All those positive samples were negative for Rickettsia typhi. Leptospira spp. was detected in one sample from a Rhinolophidae bat collected from HinHeup District. Bartonella spp. was detected in 54 bat blood samples (20.38%): 30/54 (55.56%) Hipposideridae, 20/54 (37.04%) Rhinolophidae, and 4/54 (7.41%) Vespertilionnidae.
Bat ectoparasites-bacterial screening
A total of 288 pools of 468 batflies were screened for Rickettsia spp., Leptospira spp., and Bartonella spp. Overall, 22 pools (7.64%) were positive for Rickettsia spp.: 16/22 (72.73%) Streblidae, 3/22 (13.64%) Nycteribiidae, and 3/22 (13.64%) Trombiculidae. All those positive samples were negative for Rickettsia typhi. No Leptospira spp. was detected from any samples tested. Bartonella spp. was detected in 15 pools (5.21%): 8/15 (53.33%) from Nycteribiidae, 5/15 (33.33%) from Streblidae, and 2/15 (13.33%) from Spinturnicidae.
References
Brunet-Rossinni, A.K. and G.S. Wilkinson. 2009. Methods for age estimation and the study of senescence in bats. Pp: 315-325, in Ecological and Behavioural Methods for the Study of Bats, 2nd edition (T.H. Kunz and S. Parsons, eds). The Johns Hopkins University Press, Baltimore, xvii + 901 pp.
Corbet, G.B. and J.E. Hill. 1992. The mammals of the Indomalayan region: a systematic review. Natural History Museum Publications and Oxford University Press, 488 pp.
Csorba, G., P. Ujhelyi, and N. Thomas. 2003. Horseshoe bats of the World (Chiroptera: Rhinolophidae). Alana Books, xxxii + 160 pp.
Francis, C.M. 1989. A comparison of mist nets and two designs of harp traps for capturing bats. Journal Mammal, 70: 865-870.
Francis, C.M. 2008. A field guide to the mammals of Thailand and South-east Asia. New Holland Publishers (UK) Ltd and Asia Books Co., Ltd., Bangkok, Thailand, 392 pp.
Jansen van Vuren P., M.R. Wiley, G. Palacios, N. Storm, W. Markotter, M. Birkhead, A. Kemp, J.T. Paweska. Isolation of a novel orthobunyavirus from bat flies (Eucampsipoda africana). J Gen Virol 2017 May 98(5):935-945. doi: 10.1099/jgv.0.000753. Epub 2017 May 11.
Racey, P.A. 2009. Reproductive assessment of bats. Pp: 249-264, in Ecological and behavioural methods for the study of bats, 2nd edition (T. H. Kunz and S. Parsons, eds). The Johns Hopkins University Press, Baltimore, xvii + 901 pp.
Sanchez-Seco, M.P., D. Rosario, C. Domingo, L. Hernandez, K. Valdes, M.G. Guzman, and A. Tenorio. 2005. Generic RT-nested-PCR for detection of flaviviruses using degenerated primers and internal control followed by sequencing for specific identification. J Virol Methods 126(1-2): 101-109.
Sanchez-Seco, M.P., D. Rosario, E. Quiroz, G. Guzman and A. Tenorio. 2001. A generic nested-RT-PCR followed by sequencing for detection and identification of members of the alphavirus genus. J Virol Methods 95(1-2): 153-161.
Sanchez-Seco, M.P., J.M. Echevarria, L. Hernandez, D. Estevez, J.M. Navarro-Mari, and A. Tenorio. 2003. Detection and identification of Toscana and other phleboviruses by RT-nested-PCR assays with degenerated primers. J Med Virol 71: 140-149.








